Solvation model background¶
Solvation models¶
Electrostatic and solvation models can be roughly divided into two classes ([Warshel2006], [Roux1999], [Ren2012]) explicit solvent models that treat the solvent in atomic detail and implicit solvent models that generally replace the explicit solvent with a dielectric continuum. Each method has its strengths and weaknesses. While explicit solvent models offer some of the highest levels of detail, they generally require extensive sampling to converge properties of interest. On the other hand, implicit solvent models trade detail and some accuracy for the “pre-equilibration” of solvent degrees of freedom and elimination of sampling for these degrees of freedom. Implicit solvent methods are popular for a variety of biomedical research problems.
The polar solvation energy is generally associated with a difference in charging free energies in vacuum and solvent. A variety of implicit solvent models are available to biomedical researchers to describe polar solvation; however, the most widely-used methods are currently the Generalized Born (GB) and Poisson-Boltzmann (PB) models. GB and related methods are very fast heuristic models for estimating the polar solvation energies of biomolecular structures and therefore are often used in high-throughput applications such as molecular dynamics simulations. PB methods can be formally derived from more detailed theories and offer a somewhat slower, but often more accurate, method for evaluating polar solvation properties and often serve as the basis for parameterization and testing of GB methods. Finally, unlike most GB methods, PB models provide a global solution for the electrostatic potential and field within and around a biomolecule, therefore making them uniquely suited to visualization and other structural analyses, diffusion simulations, and a number of other methods which require global electrostatic properties.
The PB equation ([Fogolari2002], [Lamm2003], [Grochowski2007], [Baker2005]) is a nonlinear elliptic partial differential equation of the form shown in the figure below which is solved for the electrostatic potential. The coefficients of this equation are directly related to the molecular structure of the system under consideration. PB theory is approximate and, as a result, has several well-known limitations which can affect its accuracy ([Grochowski2007], [Netz2000]), particularly for strongly charged systems or high salt concentrations. However, despite these limitations, PB methods are still very important for biomolecular structural analysis, modeling, and simulation. Furthermore, these limitations are currently being addressed through new implicit solvent models and hybrid treatments which extend the applicability of PB theory while preserving some of its computational efficiency. There are currently examples of both types of treatments which leverage APBS ([Azuara2006], [Chu2007], [Vitalis2004]).
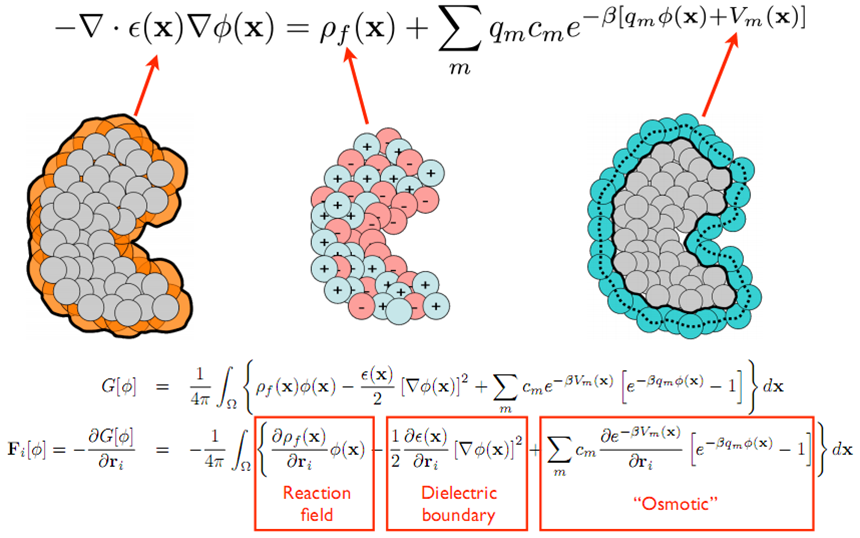
PB methods provide polar solvation energies and therefore must be complemented by non-polar solvation models to provide a complete view of biomolecular solvent-solute interactions. non-polar solvation is generally associated with the insertion of the uncharged solute into solvent. There are many non-polar solvation models available; however, work by Levy et al. [Levy2003] as well as our own research [Wagoner2006] has demonstrated the importance of non-polar implicit solvent models which include treatment of attractive solute-solvent dispersion terms. This model has been implemented in APBS and can also be easily transformed into simpler popular non-polar models (e.g., solvent-accessible surface area). While this model can be used separately from PB to analyze non-polar contributions to solvation energy, its preferred use is coupled to the PB equation through a geometric flow model [Chen2010] which treats polar and non-polar interactions in the same framework and reduces the number of user-specified empirical parameters.
Caveats and sources of error¶
Model error¶
When performing solvation calculations using APBS, it is important to keep in mind that you are using an approximate model for solvation. Therefore, your answers may contain errors related to approximations in the model. Many review articles have covered the nature of these approximations, we will stress the highlights below.
Linear dielectric response¶
The Poisson-Boltzmann equation models the solvent as a dielectric continuum that responds linearly to all applied fields. In particular, under this model, very strong fields can induce unrealistically strong polarization in the dielectric media representing the solvent and/or the solute interior. However, molecular solvents or solutes cannot support an infinite amount of polarization: they are limited by their density, their finite dipole moments, and their finite degree of electronic polarizability. Therefore, the continuum model assumption of linear dielectric response can break down in situations with strong electric fields; e.g., around nucleic acids or very highly-charged proteins.
Local dielectric response¶
The Poisson-Boltzmann equation models the solvent as a dielectric continuum that also responds locally to all applied fields. n other words, under this model, the local polarization at a point x is only dependent on the field at point x. However, molecular solvents and solutes clearly don’t obey this assumption: the variety of covalent, steric, and other non-bonded intra- and inter-molecular interactions ensures that the polarization at point x is dependent on solute-field interactions in a non-vanishing neighborhood around x. One way to limit the impact of this flawed assumption, is to model solute response as “explicitly” as possible in your continuum electrostatics problems. In other words, rather than relying upon the continuum model to reproduce conformational relaxation or response in your solute, model such response in detail through molecular simulations or other conformational sampling.
Ambiguity of dielectric interfaces and coefficient values¶
Violation of the assumptions of linear and local dielectric response in real molecular systems leads to serious ambiguity in the definition of the dielectric coefficient in the Poisson-Boltzmann equation. In particular, while the values for bulk solvent (i.e., far away from the solute) response are well-defined, all other values of the dielectric coefficient are ambiguous. In general, continuum models assume a constant low-dielectric value inside the solute and the bulk solvent value outside the solute. This assumption creates tremendous sensitivity of calculation results on the placement of the dielectric interface (usually determined by solute atomic radii) and the specific value of the internal solute dielectric. In general, errors arising from this assumption can be minimized by using internal dielectric values that are consistent with the solute atomic radii parameterization.
No specific ion-solvent or ion-solute interactions¶
Most Poisson-Boltzmann models assume that ions do not interact directly with the solvent: they are charges embedded in the same dielectric material as the bulk solvent. This assumption implies that ions experience no “desolvation” penalty as they interact with the solute surface. Additionally, most Poisson-Boltzmann models assume that ions interaction with the solute only through electrostatic and hard-sphere steric potentials. However, this assumption neglects some of the subtlety of ion-protein interactions; in particular, dispersive interactions that can possibly lead to some degree of ion specificity.
Mean field ion behavior¶
Finally, the Poisson-Boltzmann model is a “mean field” description of ionic solutions. This means that ions only experience the average influence of other ions in the system; the model neglects fluctuations in the ionic atmosphere and correlations between the ions in solution. Such correlations and fluctuations can be very important at high ionic charge densities; e.g., for multivalent ions, high ion concentrations, or the high-density ionic regions near highly-charged biomolecules.
Parameter set errors¶
Structure-based errors¶
Electrostatics calculations can be very sensitive to errors in the structure, including:
- Misplaced atoms or sidechains
- Missing regions of biomolecular structure
- Incorrect titration state assignments
Of these errors, incorrect titration states are the most common and, often, the most problematic. The software package PDB2PQR was created to minimize all of the above problems and we recommend its use to “pre-process” structures before electrostatics calculations.
Discretization error¶
The Poisson-Boltzmann partial differential equation must be discretized in order to be solved on a computer. APBS discretizes the equation in spacing by evaluating the problem coefficients and solving for the electrostatic potential on a set of grid (finite difference) or mesh (finite element) points. However, this discretization is an approximation to the actual, continuously-specified problem coefficients. Coarser discretization of coefficients and the solution reduce the overall accuracy and introduce errors into the final potential and calculated energies.
It is very important to evaluate the sensitivity of your calculated energies to the grid spacings and lengths. In general, it is a good idea to scan a range of grid spacings and lengths before starting a problem and choose the largest problem domain with the smallest grid spacing that gives consistent results (e.g., results that don’t change as you further reduce the grid spacing).
Solver and round-off error¶
APBS uses iterative solvers to solve the nonlinear algebraic equations resulting from the discretized Poisson-Boltzmann equation. Iterative solvers obtain solutions to algebraic equations which are accurate within a specified error tolerance. Current versions of APBS use a fixed error tolerance of 10-6 which implies approximately 1 part per million root-mean-squared error in calculated potentials. Such error tolerances have been empirically observed to give good accuracy in the calculated energies obtained with APBS.
However, it is important to note that the error in potential does not necessarily directly relate to the error in the energies calculated by APBS. In particular, most meaningful energies are calculated as differences between energies from several calculations. While the accuracy of each separate energy can be related to the solver error tolerance, the energy difference can only be loosely bounded by the error tolerance.
This issue is illustrated in the protein kinase ligand binding example provided with APBS as pka-lig and analyzed below.
This example demonstrates that, while the errors for each calculation remain small, the overall error in the computed energy can be very large; particularly when two different methods are compared.
| Error tolerance | Protein energy | Protein energy relative error (with respect to 10-12 tolerance) | Ligand energy | Ligand energy relative error (with respect to 10-12 tolerance) | Complex energy | Complex energy relative error (with respect to 10-12 tolerance) | Binding energy | Binding energy relative error (with respect to 10-12 tolerance) |
|---|---|---|---|---|---|---|---|---|
| 1.00E-06 | 3.01E+05 | 2.47E-08 | 1.05E+04 | 1.42E-08 | 3.11E+05 | 2.45E-08 | 8.08E+00 | 7.75E-06 |
| 1.00E-09 | 3.01E+05 | 3.19E-11 | 1.05E+04 | 1.71E-11 | 3.11E+05 | 2.45E-08 | 8.08E+00 | 2.48E-09 |
| 1.00E-12 | 3.01E+05 | 0.00E+00 | 1.05E+04 | 0.00E+00 | 3.11E+05 | 0.00E+00 | 8.08E+00 | 0.00E+00 |
Further reading¶
| [Azuara2006] | Azuara C, Lindahl E, Koehl P, Orland H, and Delarue M, PDB_Hydro: incorporating dipolar solvents with variable density in the Poisson-Boltzmann treatment of macromolecule electrostatics. Nucleic Acids Research, 2006. 34: p. W38-W42. |
| [Baker2005] | Baker NA, Biomolecular Applications of Poisson-Boltzmann Methods, in Reviews in Computational Chemistry, Lipkowitz KB, Larter R, and Cundari TR, Editors. 2005, John Wiley and Sons. |
| [Chen2010] | Chen Z, Baker NA, Wei GW. Differential geometry based solvation model I: Eulerian formulation, J Comput Phys, 229, 8231-58, 2010. |
| [Chu2007] | Chu VB, Bai Y, Lipfert J, Herschlag D, and Doniach S, Evaluation of Ion Binding to DNA Duplexes Using a Size-Modified Poisson-Boltzmann Theory. Biophysical Journal, 2007. 93(9): p. 3202-9. |
| [Fogolari2002] | Fogolari F, Brigo A, and Molinari H, The Poisson-Boltzmann equation for biomolecular electrostatics: a tool for structural biology. Journal of Molecular Recognition, 2002. 15(6): p. 377-92. |
| [Grochowski2007] | (1, 2) Grochowski P, lstrok A, and Trylska J, Continuum molecular electrostatics, salt effects and counterion binding. A review of the Poisson-Boltzmann theory and its modifications. Biopolymers, 2007. 89(2): p. 93-113. |
| [Lamm2003] | Lamm G, The Poisson-Boltzmann Equation, in Reviews in Computational Chemistry, Lipkowitz KB, Larter R, and Cundari TR, Editors. 2003, John Wiley and Sons, Inc. p. 147-366. |
| [Levy2003] | Levy RM, Zhang LY, Gallicchio E, and Felts AK, On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. Journal of the American Chemical Society, 2003. 125(31): p. 9523-30. |
| [Netz2000] | Netz RR and Orland H, Beyond Poisson-Boltzmann: Fluctuation effects and correlation functions. European Physical Journal E, 2000. 1(2-3): p. 203-14. |
| [Ren2012] | Ren P, Chun J, Thomas DG, Schnieders M, Marucho M, Zhang J, Baker NA, Biomolecular electrostatics and solvation: a computational perspective. Quarterly Reviews of Biophysics, 2012. 45(4): p. 427-491. |
| [Roux1999] | Roux B and Simonson T, Implicit solvent models. Biophysical Chemistry, 1999. 78(1-2): p. 1-20. |
| [Vitalis2004] | Vitalis A, Baker NA, McCammon JA, ISIM: A program for grand canonical Monte Carlo simulations of the ionic environment of biomolecules, Molecular Simulation, 2004, 30(1), 45-61. |
| [Wagoner2006] | Wagoner JA and Baker NA, Assessing implicit models for nonpolar mean solvation forces: the importance of dispersion and volume terms. Proceedings of the National Academy of Sciences of the United States of America, 2006. 103(22): p. 8331-6. |
| [Warshel2006] | Warshel A, Sharma PK, Kato M, and Parson WW, Modeling electrostatic effects in proteins. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics, 2006. 1764(11): p. 1647-76. |